
The human genome is composed of approximately 3 billion letters.
Eighteen years ago, the world celebrated the publication of the complete human genome by the Human Genome Project (HGP).
The 15-year project, whose goal was to decipher the code used in our genetic makeup and create an encyclopedia of human genes, had finally accomplished its task. Or more precisely, it had almost completed the work. The media and the public largely overlooked one crucial detail in that publication: the HGP had decoded only the euchromatic genome – the lightly compressed portion of the chromosomes. The heterochromatic genome – the tightly packed portion of the chromosomes – remained undeciphered.
Chromatin is the material that makes up chromosomes. It is a complex formed by DNA and proteins that helps pack long DNA molecules into cells.
There are two categories of chromatin: euchromatin and heterochromatin. The former is characterized by lightly packed DNA that allows proteins to transcribe, or copy, DNA into RNA, which binds to the DNA more easily as a result. The latter is a tightly packed, supercoiled DNA that does not allow gene transcription.
The HGP used world-class sequencing technology, but even that was not sufficient at the time to properly sequence the heterochromatic region of the human genome. So, they focused on the euchromatic region, which comprises about 92% of the human genome, and left the remaining 8% to the future. In March 2022, a consortium of researchers took a giant step toward that future by publishing the most accurate and complete human genome ever produced.
Since the first publication of a human genome, some regions have remained inaccessible for nearly two decades.
The challenges of sequencing the complete human genome revolve around two main factors: its enormous size – our genome consists of 3 billion letters; and its complexity – some regions are difficult to access and consist of highly repetitive sequences that are difficult to assemble.
A global team of researchers has now published a series of six articles that closed most of the gaps in sequencing the human genome. The developments reveal the points most susceptible for gene evolution – with configurations that promote the emergence of new genetic variations – in the newly sequenced regions and spur researchers to keep working towards finally assembling a truly complete human genome.
.")
Fig. Each bar represents one of the 23 chromosomes of the genome sequenced by the T2T Consortium. The previously missing sequences that the T2T Consortium resolved are in red.
When the HGP published the euchromatic human genome in 2004, sequencing equipment and assembly software were unable to properly process some regions of DNA with highly repetitive base sequences.
At the time, the technology struggled to avoid skipping repeats and to figure out how the bases were connected.
As researchers continued to improve the technology, Dr. Karen Miga, a geneticist and assistant professor at the University of California, Santa Cruz, and Dr. Adam Phillippy, a bioinformatician at the National Human Genome Research Institute, teamed up to fill in the gaps in the human genome. Using new sequencing technologies capable of reading much longer stretches of DNA at once, which makes the job of stitching sequences together easier, they succeeded in sequencing the X chromosome from end to end in 2019.
The breakthrough inspired dozens of other researchers to join the cause and led to the emergence of a global task force devoted to assembling the first complete human genome. Dr. Miga, Dr. Phillippy, and Dr. Evan Eichler, a geneticist at the University of Washington, co-led the group, called the Telomere-to-Telomere (T2T) Consortium. The consortium’s name hints at its goal of sequencing the human genome from one end to the other – telomeres are the regions at the tips of chromosomes, made up of repeated sequences of non-coding DNA that protect chromosomes from damage.
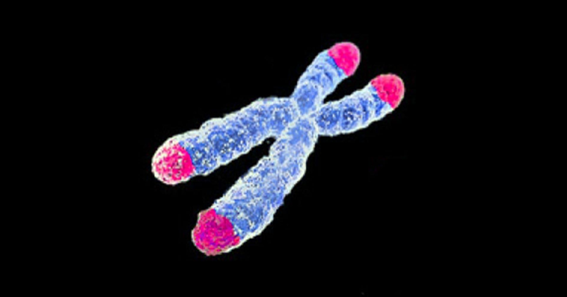
Chromosomes are X-shaped structures that pack the DNA inside cells. The tips of the chromosomes (in pink) are called telomeres and the intersection between the legs of the structure is called centromere.
The T2T team used genetic material from a cell line derived from a molar pregnancy – a type of failed pregnancy in which a sperm penetrates an egg that lacks its own chromosome.
This phenomenon generates a cell that has only the genetic material of the sperm and cannot develop into an embryo but can replicate, especially if it carries an X chromosome.
As that cell self-replicates, both members of the 23 pairs of chromosomes are identical. This characteristic is very helpful when sequencing the genome because it means that the differences between the chromosomes of the parents no longer have to be considered when eliminating gaps. In addition, because the mole DNA used by the team has no Y chromosome, they used DNA donated by a 51-year-old biologist at Harvard University to sequence that specific chromosome.
The consortium has disentangled previously indecipherable portions of the genome, providing deeper insight into the human genome. Innovations include sequencing of telomeres, centromeres (which are the dense knobs that typically mark the center of the chromosome), and the short arms of the five chromosomes in which the centromeres are shifted toward one end.
The result of mapping and sequencing some 200 million bases that make up more than 1,900 genes once again highlights the complexity of the human genome and opens up potential opportunities for the development of medical treatments as well as for our overall knowledge of the human body and evolutionary history.
In these previously unmapped regions of the genome, the researchers found duplicated regions and mobile elements, which are genetic material from viruses that were incorporated into our DNA at some point during evolution. They also found that the size of duplicated regions in centromeres varies widely, although our current knowledge suggests that their function does not.
The short arms of chromosomes also revealed some exciting information about the human makeup.
These short arms are packed with copies of various types of repetitive DNA, including duplicated regions, mobile elements, and eventually multiple copies of the genes that code for the RNA responsible for making ribosomes. Dr. Phillippy explains that these findings suggest that the short arms are likely hotspots for evolution because all these gene copies are lying around freely to mutate and develop new functions.
The T2T data will provide a reference for mapping variations in these previously unmapped regions that are potentially of great medical importance.
Chemical changes in complex, repetitive regions of DNA may be linked to some types of diseases, and some neurological and developmental disorders are related to variations in the number of copies of certain sequences in DNA. This catalog of repetitive DNA can contribute to a better understanding of the mechanisms behind such conditions and, consequently, to the development of strategies to circumvent and treat them.

The next step of the T2T Consortium will be to expand their sample to capture the genetic diversity of the human genome.
However, the researchers emphasize that the work is far from complete.
Even though the recent contributions of the T2T consortium have been a giant leap, there are still hundreds of gaps in the genome, and they still have not succeeded in sequencing a chromosome from end to end.
Additionally, they have only worked with genetic material from two people – the anonymous father of the mole and the donor of the Y chromosome. So there is still a long way to go. To create a representative reference guide to the human genome, the next steps of the T2T Consortium will be to sequence the genomes of more people with diverse genetic backgrounds so that our knowledge of the human genome adequately covers our genetic diversity.
This step is already underway as part of the Human Pangenome Reference Consortium.
The T2T team has already sequenced dozens of other genomes and aims to do so with 350 genomes of diverse ancestry. As the researchers move forward, they hint at their ultimate goal: the day when sequencing technology will be so advanced that we will have a nearly complete reference guide to the human genome, as well as the ability to sequence any genome on demand. That day will open a new chapter for humanity’s ability to deal with genetic disorders, from predicting to treating them.

